2025年2月28日是第18個(gè)國(guó)際罕見(jiàn)病日,今年主題是“More than you can imagine”,,意為“不止罕見(jiàn)”,。希望通過(guò)罕見(jiàn)病科普宣教,讓罕見(jiàn)病被更多人看見(jiàn),、被關(guān)注,、被了解,關(guān)愛(ài)罕見(jiàn)病患者,。
您是否也認(rèn)為罕見(jiàn)病離我們很遙遠(yuǎn),?
您是否也認(rèn)為家族成員沒(méi)有遺傳病,自己孩子絕不會(huì)患罕見(jiàn)???
事實(shí)上,全球范圍內(nèi)約有3億人正遭受罕見(jiàn)病的折磨,,而我國(guó)每年新增的罕見(jiàn)病患者數(shù)量超過(guò)20萬(wàn),。這些罕見(jiàn)病的癥狀往往與普通疾病相似,因此極易導(dǎo)致誤診或漏診的情況發(fā)生,。我國(guó)罕見(jiàn)病患者的平均確診時(shí)間約為4~5年,,部分患者甚至需要更長(zhǎng)時(shí)間。罕見(jiàn)病約50%~60%為隱性遺傳,,基因的變異可能來(lái)自健康的父母,。
每個(gè)學(xué)科都有很多罕見(jiàn)病,這些疾病的病因復(fù)雜多樣,,可能涉及遺傳因素,、基因突變、代謝異常等,。今日,,讓我們來(lái)認(rèn)識(shí)和了解幾種典型的骨科罕見(jiàn)病。由于這些骨科罕見(jiàn)病的發(fā)病率極低,,每一種疾病的患者數(shù)量都相對(duì)較少,,因此它們常被形象地稱為“孤兒病”。被國(guó)家列入罕見(jiàn)病名錄的骨科疾病包括脆骨病,、先天性脊柱側(cè)彎,、馬凡綜合癥和腓骨肌萎縮癥等。
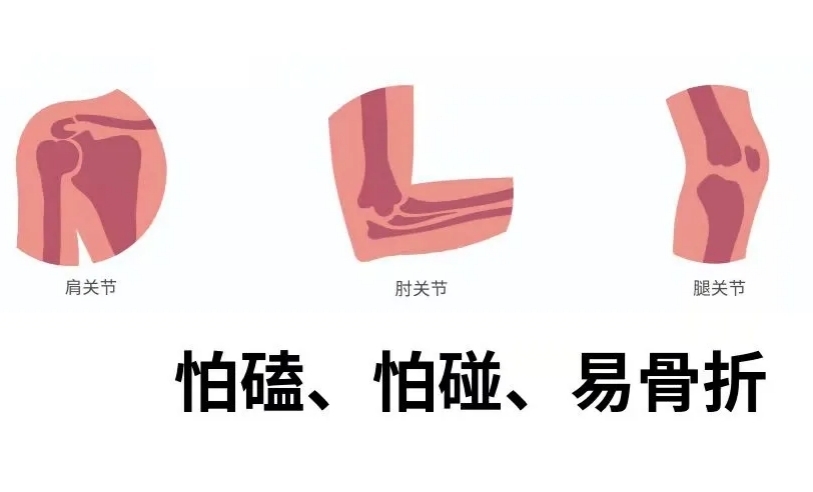
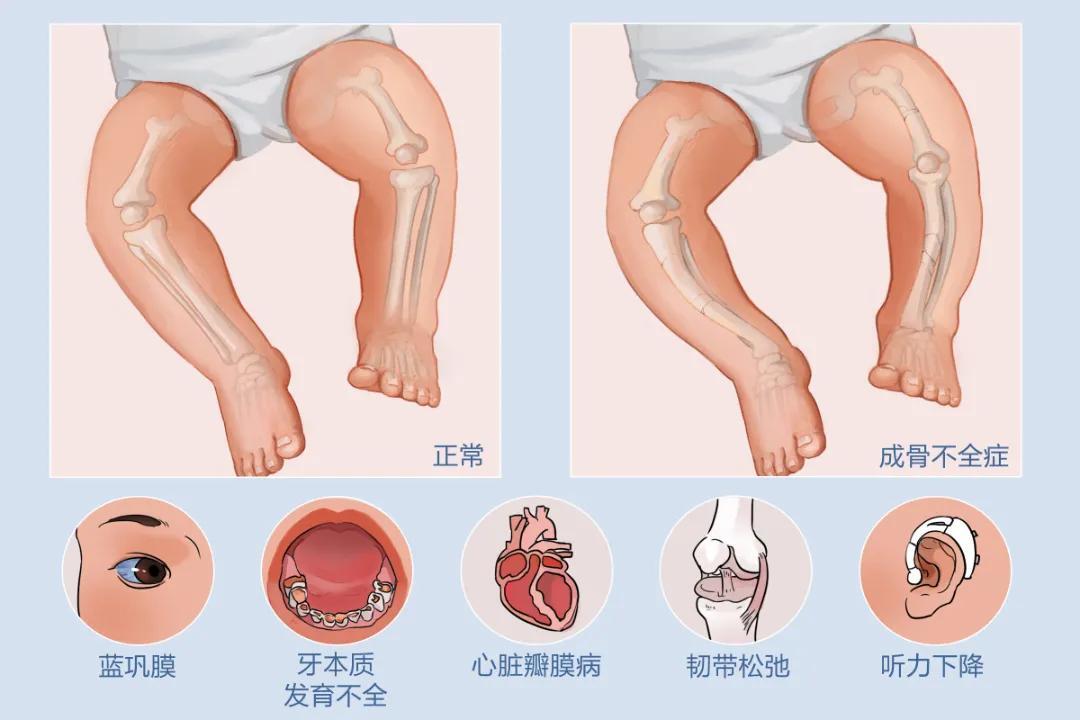
成骨不全癥(OI)——又稱“脆骨病”或“瓷娃娃病”,,是一種因基因缺陷導(dǎo)致膠原蛋白合成異常的遺傳性骨病 ,?;颊吖趋来嗳酰缤善靼闳菀灼扑?,輕微碰撞,,甚至日常活動(dòng)都可能引發(fā)骨折,。據(jù)統(tǒng)計(jì),,每1萬(wàn)-2萬(wàn)名新生兒中就有1例發(fā)病。由于頻繁骨折,,患者常伴有骨骼畸形,、身材矮小等問(wèn)題,嚴(yán)重影響生活質(zhì)量,。
主要有哪些表現(xiàn),?
兒童期,很多患者從嬰兒時(shí)期就頻繁骨折,,骨折次數(shù)隨著年齡增長(zhǎng)不斷增加,。同時(shí),他們的身材往往比同齡人矮小,,肢體發(fā)育也可能出現(xiàn)畸形,。
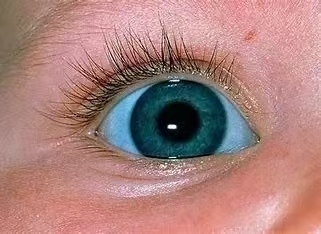
而且,由于鞏膜變得異常薄弱,,大部分患者的眼睛會(huì)呈現(xiàn)出藍(lán)色或灰色,,就像深邃的寶石,這也是成骨不全癥的典型特征之一,。
成年后,,除了骨折風(fēng)險(xiǎn)持續(xù)存在,還可能出現(xiàn)牙齒發(fā)育異常,,比如牙本質(zhì)發(fā)育不全,,牙齒容易磨損、折斷,。此外,,聽(tīng)力下降也是常見(jiàn)問(wèn)題,給患者的生活帶來(lái)諸多不便,。
引發(fā)原因是什么,?
這種病主要由遺傳因素導(dǎo)致,多數(shù)為常染色體顯性遺傳,,即父母一方患病,孩子就有50%的概率遺傳到致病基因,。然而,,仍有一部分病例源自基因突變,,這種突變是自發(fā)的,即便父母雙方均無(wú)相關(guān)家族病史,,孩子仍可能罹患此病,。
目前能治療和預(yù)防嗎?
目前,,脆骨病無(wú)法完全治愈,,但通過(guò)積極治療能改善癥狀。雙膦酸鹽類藥物可以增加骨密度,,降低骨折風(fēng)險(xiǎn),;外科手術(shù)如髓內(nèi)釘固定術(shù),能幫助矯正骨骼畸形,,改善肢體功能,。對(duì)于有家族遺傳史的家庭,備孕前進(jìn)行遺傳咨詢和基因檢測(cè)非常重要,,可以評(píng)估生育風(fēng)險(xiǎn),。

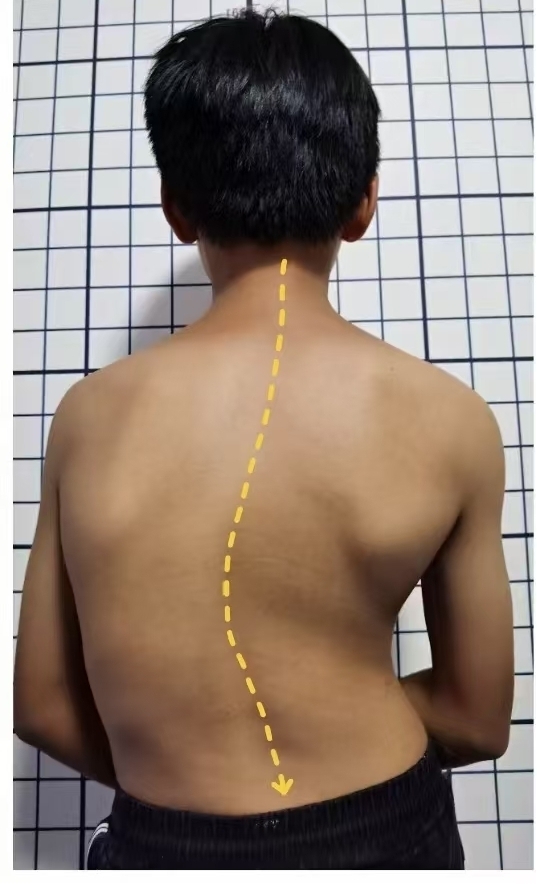
先天性脊柱側(cè)彎——是指先天性椎體異常而引起的脊柱側(cè)凸。這種畸形出生后即發(fā)病,,因而患者出現(xiàn)畸形較特發(fā)性脊柱側(cè)凸早,。由于先天性脊柱側(cè)凸往往在早期即發(fā)病,這使得患者往往難以獲得及時(shí)且最佳的治療,。由于形成的彎曲易于進(jìn)展,,并且患者仍有較長(zhǎng)的生長(zhǎng)期,所以容易產(chǎn)生較嚴(yán)重的畸形,。
目前尚無(wú)法得知先天性脊柱側(cè)彎的真正發(fā)病原因,,大多數(shù)學(xué)者認(rèn)為環(huán)境、遺傳,、維生素缺乏,、化學(xué)物質(zhì)、有毒物質(zhì)等諸多因素中的一種或幾種均在脊柱生長(zhǎng)發(fā)育不同階段參與及影響脊柱側(cè)彎的形成,。其治療可分為兩大類,,即非手術(shù)治療和手術(shù)治療。常見(jiàn)的非手術(shù)治療方法包括理療,、體操療法,、石膏、支具等,,但最主要和最可靠的方法是支具治療,。一旦先天性脊柱側(cè)彎出現(xiàn)明顯進(jìn)展,則應(yīng)盡早手術(shù)治療,,通常3至5歲是一個(gè)比較好的手術(shù)時(shí)機(jī),。
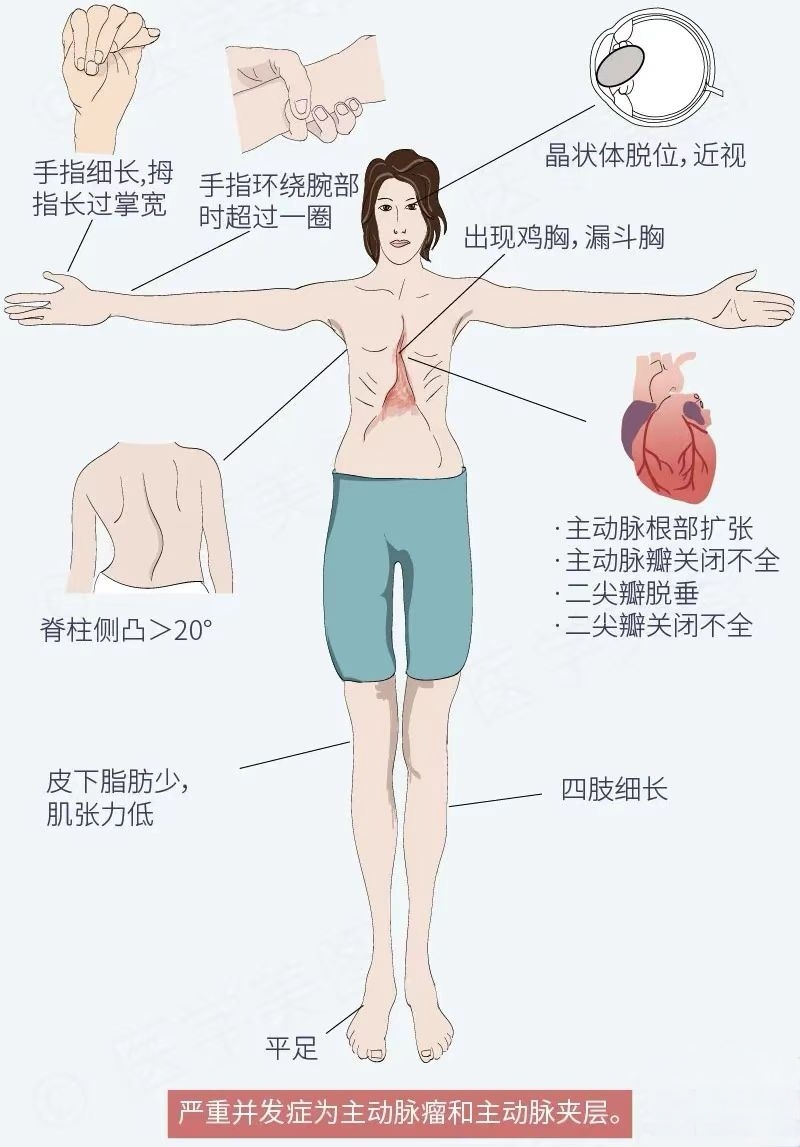
馬凡綜合征——又稱“天才病”和“巨人殺手”,,是一種遺傳性結(jié)締組織疾病,因累及骨骼使手指細(xì)長(zhǎng),,呈蜘蛛指(趾)樣,,故又稱為蜘蛛指(趾)綜合征。它屬先天性遺傳性疾病,,發(fā)病率約為1/3,000-1/5,000,,發(fā)病率低但死亡率極高,該病致病基因攜帶者有一半的概率將其傳給下一代,。大多數(shù)馬凡氏綜合征患者有家族史,,但同時(shí)又有15~30%的患者是由于自身基因突變導(dǎo)致的,患者自然生存壽命平均僅為32歲,。
此病以骨骼,、眼及心血管三大系統(tǒng)為主要特征,患者四肢,、手指,、腳趾細(xì)長(zhǎng)不勻稱,身高明顯超出常人,?;加旭R凡氏病的病人,往往并發(fā)心血管疾病,,且多數(shù)人在30至50歲因心血管疾病而不幸離世,。該病同時(shí)可能影響其他器官,包括肺,、眼,、硬脊膜、硬顎等,。
馬凡綜合征已經(jīng)奪走了多名著名大個(gè)子運(yùn)動(dòng)員的生命,,1986年,31歲的美國(guó)女排名將海曼在比賽中猝死球場(chǎng),,死亡原因正是馬凡綜合征引起的主動(dòng)脈夾層破裂,。1984年,身高2.17米的中國(guó)男籃運(yùn)動(dòng)員韓朋山猝死,,時(shí)年28歲,;2001年,身高2.04米的四川排球運(yùn)動(dòng)員朱剛猝死,,那年他剛滿30歲,;2009年,身高2.12米的沈陽(yáng)籃球運(yùn)動(dòng)員武強(qiáng)突發(fā)心臟病去世,年僅23歲,;2012年,,曾經(jīng)被譽(yù)為“小姚明”的原遼寧男籃隊(duì)員張佳迪突發(fā)心臟病去世,年僅24歲,。
馬凡綜合征是一種顯性遺傳病,對(duì)于有疑似馬凡氏綜合征臨床表現(xiàn)的,,應(yīng)該先排查家族史,,并進(jìn)行相關(guān)檢查,包括基因檢測(cè),、心電圖,、超聲心動(dòng)圖、掌骨及胸部X線片,,必要時(shí)行胸腹動(dòng)脈核磁檢查,,爭(zhēng)取做到早發(fā)現(xiàn)早治療。對(duì)于有健康生育需求的夫婦,,可以通過(guò)婚前檢查遺傳咨詢,、產(chǎn)前檢查和遺傳病的早期治療,避免下一代遺傳馬凡綜合征和基因變異的發(fā)生,。
這些骨科罕見(jiàn)病,,由于其特殊性,至今仍大多缺乏有效的治療手段,,患者往往面臨長(zhǎng)期病痛和高昂醫(yī)療費(fèi)用,。但隨著醫(yī)學(xué)研究的不斷進(jìn)步,新的治療手段和藥物正在研發(fā)中,。他們大多數(shù)是基因疾病,,也就是從母體帶出來(lái)的,因此,,重視產(chǎn)前篩查,,從源頭進(jìn)行防控,是預(yù)防和應(yīng)對(duì)罕見(jiàn)病最經(jīng)濟(jì)有效的健康策略,。
希望大家能關(guān)注這些罕見(jiàn)病群體,,讓更多人了解骨罕見(jiàn)病的癥狀、診斷和治療,,不僅能減少對(duì)患者的誤解和歧視,,還能使更多患者得到早期診斷和治療。全社會(huì)共同努力,,提高對(duì)骨罕見(jiàn)病的認(rèn)知,,加強(qiáng)醫(yī)療支持,給予他們更多理解和關(guān)愛(ài),,為這些 “折翼的天使”撐起一片希望的天空,。(骨科一區(qū)宋漢華,、李香芙供稿)


